Research Article
Splenic Transcriptome Profiling of the Patients with Post-Hepatitis B Liver Cirrhosis and Portal Hypertension
Lingxiang Yu #, Xiaodong Guo #, Zhiwei Li, Shaogeng Zhang *
Department of Hepatobiliary Surgery, 302 Hospital of PLA, Beijing 100039, PR China.
# These authors contributed equally to this paper and should be regarded as co-first author.
* Corresponding author. E-mail: ylx302@163.com
Received: Sep. 19, 2016; Accepted: Sep. 20, 2016; Published: Sep. 28, 2016.
Citation: Lingxiang Yu, Xiaodong Guo, Zhiwei Li and Shaogeng Zhang, Splenic Transcriptome Profiling of the Patients with Post-Hepatitis B Liver Cirrhosis and Portal Hypertension. Nano Biomed. Eng., 2016, 8(3): 184-189.
DOI: 10.5101/nbe.v8i3.p184-189.
Abstract
The infection with hepatitis B virus is the common cause of liver cirrhosis, and portal hypertensionis is one of the main causes of morbidity and mortality in patients with cirrhosis, which has attracted extensive attention for its fatal complications worldwide. By comparing the expression pattern to normal spleen tissues through RNA-Seq technology, we identified a list of protein-coding genes that were expressed differentially between patient and normal samples. Especially the secretory cytokines, including chemokine activity-related genes and immediate early response genes, were significantly upregulated in spleen samples. The results and genes identified in this study revealed the similar expression pattern in human samples to that reported in murine model, which further support the thesis that the spleen could take a harmful role and provide a negative impact in this situation due to inducing chemokine and transcription factor.
Keywords: Hepatitis B; Liver cirrhosis; Portal hypertension; Transcriptome sequencing
Viral hepatitis afflicts hundreds of millions of people. The infection by hepatitis-causing viruses can lead to significant morbidity and mortality [1]. Especially hepatitis B (HBV), discovered more than half a century ago, infects estimated 2 billion people including more than 350 million chronical carriers of HBV, and accouts for 786, 000 deaths worldwide annually from cirrhosis, liver failure, and hepatocellular carcinoma [2-4]. Besides the clinical outcomes of HBV infection mentioned above, the imbalance of humoral responses to HBV can generate circulating immune complexes, which may cause extrahepatic manifestations: glomerulonephritis and vasculitis,etc.[5]. In sub-Saharan Africa and most parts of Asia, the infection with hepatitis B virus is the most common cause of liver cirrhosis, which is histologically characterised by diffuse nodular regeneration surrounded by dense fibrotic septa with subsequent parenchymal extinction and collapse of liver structures, as well as distortion of hepatic vascular architecture [6]. Depending on the severity of the cirrhosis, it leads to hemodynamic disorders including portal hypertension and hyperdynamic circulation, which are the main causes of morbidity and mortality in patients with cirrhosis [7-9]. Technological advances in the sequencing field enable in-depth characterization of the transcriptome. The transcriptome, namely the complete collection of RNA molecules expressed in a specific cell or tissue, includes mRNA and numerous non-coding RNAs, such as ribosomal RNA, transfer RNA, small nuclear RNA, small nucleolar RNA, micro RNA and so on [10-12]. Characterization of the transcriptome is the key to understanding the functional elements of the genome, revealing the molecular constituents of cells and tissues, so much so that many diseases can be understood in depth. RNA-Seq is a recently developed method for transcriptome profiling that uses deep-sequencing technologies, which have already changed our view of the extent and complexity of eukaryotic transcriptomes. Here we presented a study that used RNA-Seq to characterize the splenic transcriptome profile of coding genes in a group of patients with post-hepatitis B liver cirrhosis and portal hypertension. By comparing the expression pattern to normal spleen tissues, we identified a list of protein-coding genes that were expressed differentially between patient and normal samples, especially the secretory cytokines expressed differentially in spleen, which could be helpful for further functional dissection or the clarification of etiopathogenesis.
Materials and Methods
Spleen tissue samples
Eleven patients with advanced liver cirrhosis (6 males, 5 females; mean age: 55.4 years; positive HBV infection, Child–Pugh grade B/C: 8/3) who underwent splenectomy at our department of No.302 PLA hospital between May 2010 and April 2014 were enrolled in this study. Patients with Child–Pugh grade A cirrhosis were excluded from this study. Two spleen samples from patients underwent splenectomy for ruptured spleen were taken as normal reference samples. After obtaining the approval of Human Research Protective Committee of No.302 PLA hospital and informed consents from the patients, fresh tissue samples were taken during surgical resection, snap-frozen on dry ice and stored at −80℃. Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and the integrity of RNA was assessed by visualization of the 18S and 28S RNA on electrophoresis in agarose gel after ethidium bromide staining (Sigma–Aldrich , St. Louis, MO, USA).
RNA-Seq of patients’samples
The preparation of mRNAs library were carried out with NEBNext Kit for Illumina (New England Biolabs, Ipswich, MA, USA). Briefly, mRNA were enriched by magnetic oligo-dT beads and randomly fragmented. First-strand cDNA synthesis was performed using random hexamers and reverse transcriptase. After ligated with Adaptor Mixs and amplified with PCR, the cDNAs were sequenced by whole-transcriptome sequencing on the Illumina HiSeq 2000 platform.
Identification of differentially expressed transcripts
To estimate the transcription level of protein-coding genes, we downloaded the gene set of the Reference Sequence (RefSeq) genes under current human genome assembly (hg19) from http://genome.ucsc.edu/ and mapped RNA-Seq reads back to the reference genome (hg19) by TopHat (http://ccb.jhu.edu/software/tophat/index.shtml), which was used to align RNA-Seq reads to mammalian-sized genomes using the ultra high-throughput short read aligner Bowtie and then analyze the mapping results to identify splice junctions between exons. The relative abundances of each transcript were estimated by counting the number of reads mapped to the genomic location of that transcript. To analyse the differentially expressed genes between patients’ and normal reference samples, we used t-test with unequal variation. The supervised clustering analysis Hierarchical clustering of gene expression was performed by R and the gene functions and pathways were annotated using the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resource.
Results
Data summary
The splenic RNA-Seq transcriptome dataset contains 10,645,928 clean reads and 12,915,031 clean reads in patients’ samples and normal reference samples respectively. Total mapped reads are 9,678,864 and 11,789,736 for patients’ samples and normal reference samples respectively, with the mapped percentage from 90.92% to 91.29%. There is no significant discrepancy observed for alignment percentage between those samples, so all the data were used in further differentially expression analysis.
Expression analysis
To evaluate the expression level of transcripts, the TopHat-processed result was analysed with Cufflinks (http://cole-trapnell-lab.github.io/cufflinks/), which could assemble transcripts, estimate their abundances, and tests for differential expression and regulation in RNA-Seq samples based on a statistical model of paired-end sequencing experiments, and give a fragment per kilobase of exon per million (FPKM) value for each transcript. According to the results, 228 differentially expressed transcripts were either up-regulated or down-regulated in patients’ samples compared to normal reference samples (Fig.1). The differentially expressed genes were ranked by the log2 Ratio (case vs control) and only top 10 up- or down-regulated genes were listed in Table.1.
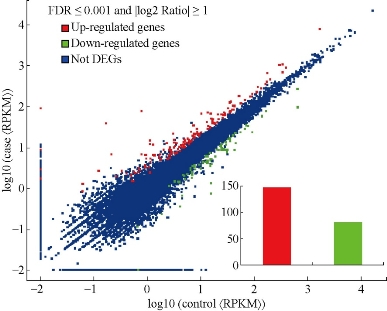
Fig.1. The differentially expressed genes (DEG) in the spleen of patients with post-hepatitis B liver cirrhosis and portal hypertension compared to normal reference samples. The inset indicates the number of DEG.
Table 1 The top 10 differentially expressed genes were ranked by the log2 Ratio.
|
log2 Ratio (case/control) |
Up or Down Regulation (case/control) |
Symbol |
Description |
|
13.12056 |
Up |
ANKRD1 |
ankyrin repeat domain 1 (cardiac muscle) |
|
9.757054 |
Up |
MT1M |
metallothionein 1M |
|
8.172454 |
Up |
C21orf128 |
chromosome 21 open reading frame 128 |
|
7.813032 |
Up |
XIST |
X inactive specific transcript (non-protein coding) |
|
7.444275 |
Up |
GYPA |
glycophorin A (MNS blood group) |
|
6.623361 |
Up |
MTRNR2L6 |
MT-RNR2-like 6 |
|
4.951984 |
Up |
NTS |
neurotensin |
|
4.445024 |
Up |
LOC284889 |
uncharacterized LOC284889 |
|
4.367021 |
Up |
MAGEC1 |
melanoma antigen family C, 1 |
|
3.741417 |
Up |
CSMD2 |
CUB and Sushi multiple domains 2 |
|
-6.07967 |
Down |
CSMD1 |
CUB and Sushi multiple domains 1 |
|
-5.16046 |
Down |
FAM9C |
family with sequence similarity 9, member C |
|
-4.38787 |
Down |
SIGLEC14 |
sialic acid binding Ig-like lectin 14 |
|
-3.56372 |
Down |
SIGLEC12 |
sialic acid binding Ig-like lectin 12 (gene/pseudogene) |
|
-3.50334 |
Down |
KIR3DX1 |
killer cell immunoglobulin-like receptor, three domains, X1 |
|
-3.01236 |
Down |
C4B_2 |
complement component 4B (Chido blood group), copy 2 |
|
-2.7571 |
Down |
BPI |
bactericidal/permeability-increasing protein |
|
-2.75462 |
Down |
HLA-DRB5 |
major histocompatibility complex, class II, DR beta 5 |
|
-2.50334 |
Down |
FREM2 |
FRAS1 related extracellular matrix protein 2 |
|
-2.31748 |
Down |
HSPA1B |
heat shock 70kDa protein 1B |
Functional and pathway analysis of differentially expressed RefSeq genes
To better understand the differentially expressed RefSeq genes, DAVID tools were used to annotate these genes. By the analysis of gene ontology (GO), 26 terms of biological process were found among these RefSeq genes and the most significant terms were cellular process, metabolic process, regulation of biological process, biological regulation, signaling and response to stimulus, etc. (Fig.2). In comparison, there were 10 terms of molecular function and 10 terms of cell component found in these RefSeq genes, and the most significant ones are related to binding, cell and cell part respectively (Fig.2). Besides, we further investigated pathway annotations of the differentially expressed genes; top 20 statistics of pathway enrichment for control VS case were shown in Fig.3. Notably, 13 differentially expressed genes with pathway annotation were identified in Cytokine-cytokine recept or interaction pathway by Kyoto Encyclopedia of Genesand Genomes (KEGG) and classified into Chemokines CXC subfamily, Chemokine CC subfamily, PDGF (Platelet Derived Growth Factor) family, TNF (Tumor Necrosis Factor) family and IL-1 (interleukin-1) family, respectively. Compared with normal reference samples, there were 8 cytokines up- or down-regulated in patients’ spleens.
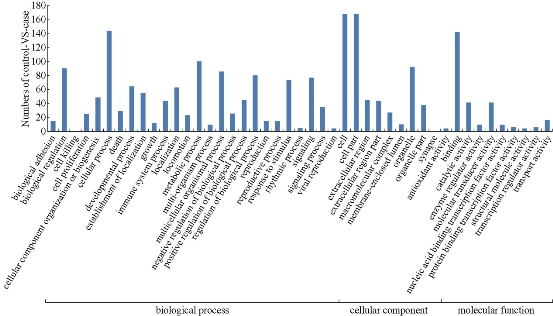
Fig.2. The analysis of gene ontology of the differentially expressed genes.
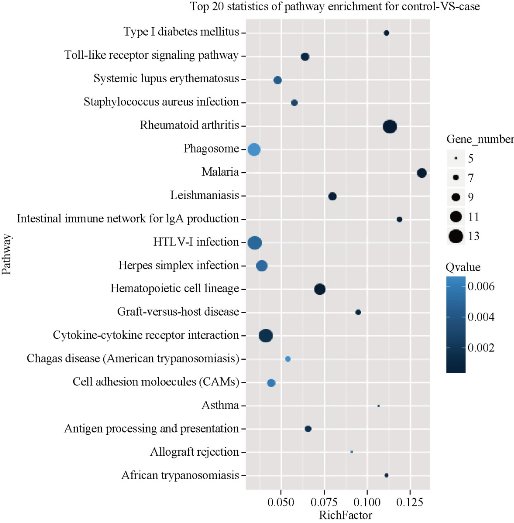
Fig.3.Top 20 statistics of pathway enrichment for the differentially expressed genes.
Discussion
Cirrhotic portal hypertension has attracted extensive attention for its fatal complications for hundreds of years [13,14]. Its consequences include bleeding from gastroesophageal varices, portal hypertensive gastropathy, ascites, spontaneous bacterial peritonitis, hepatorenal syndrome, hepatopulmonary/portopulmonary syndromes and hepatic encephalopathy, which represent the first cause of death and liver transplantation in cirrhotic patients. The primary cause of portal hypertension is the significantly increased hepatic vascular resistance (HVR) to portal blood flow, which was attributed to distortion of the liver architecture owing to cirrhosis [14-17]. Besides, the progressive splanchnic vasodilatation increases the portal blood flow, which further aggravates the state of portal hypertension [14,18]. Although not fully elucidated, it is generally accepted that portal hypertension is involved by many etiologic factors including increased vasodilatory substance release, dys-regulated response to vasoconstrictors, changes of vascular contractile signaling, vascular remodeling, and angiogenesis [13]. Therefore, it would probably be an alternative way to explore the changes of vasoactive or some other cytokines in cirrhotic portal hypertension for better elucidation of its pathogenesis. It is well recognized that the spleen is one of the abdominal organs connected with the portal system. Its major functions cover the clearance of erythrocytes and blood-borne microorganism, recycling of iron, elicitation of immunity as well as supplement of erythrocytes under hemorrhagic state [19]. Although its importance in hemopoiesis and immunity, the excision of spleen was reported to protect hepatocytes and improve hepatic functions in the settings of portal hypertensive cases or other cases with excessive portal vein flow [20-23]. The exact mechanism was still unclear. The related researches mainly focused on what happened in liver, seldom in spleen. Recently, the gene profile in the spleen after experimental hepatectomy in mice has shown that the significant differential expression of numerous inflammatory-related genes occurred in the spleen of mice after 90% hepatectomy [20]. However, the data from clinical samples have been not available to further support the thesis until now. In our work, the results revealed that the genes of the chemokine activity showed significant different patterns between the two groups. The up- or down-regulated genes were the same as the previous report in mice experiment, which meant that there was acute phase response involved in the spleen of patients with post-hepatitis B liver cirrhosis and portal hypertension [20]. In addition, early growth response-1 (EGR1), one of the zinc-finger transcription factors, was also found to be upregulated in the spleen samples of those patients. In the previous work, EGR1 was reported to be a master switch coordinating upregulation of divergent gene families related to liver is chemia-reperfusion injury including IL-1β, Cxcl2 (MIP-2), tissue factor, and plasminogen-activating inhibitor 1 [20-24]. These results showed that there were similar changes occurred in human spleen samples under the state of cirrhotic portal hypertension like what happened in mice hepatectomy experiment. In conclusion, we report the splenic transcriptome profile of coding genes in a group of patients with post-hepatitis B liver cirrhosis and portal hypertension. Although we do not report compelling evidence owing to the lack in the data of cytokine and transcription factor in portal flow, the results and genes identified in this study revealed the similar expression pattern in human samples to that in murine model, which further support the thesis that the spleen could take a harmful role and provide a negative impact in this situation due to inducing chemokine and transcription factor.
Conflict of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This project is supported by 302 Hospital of PLA.
References
Copyright© 2016 Lingxiang Yu, Xiaodong Guo, Zhiwei Li and Shaogeng Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
|
Nano Biomedicine and Engineering. Copyright © Shanghai Jiao Tong University Press |
 |